Generator
Overview
In this study, we introduced Generator, a collection of generative genomic foundation models utilizing the transformer decoder architecture, trained on expansive DNA datasets derived from the RefSeq database. Our evaluations demonstrate that the Generator consistently achieves state-of-the-art performance across a wide spectrum of benchmarks, including Genomic Benchmarks, NT tasks, and our newly proposed Gener tasks.
Beyond benchmark performance, the Generator adheres to the central dogma of molecular biology, accurately generating protein-coding DNA sequences that produce proteins structurally analogous to known families. Moreover, the Generator showcases significant promise in sequence optimization, particularly in the design of enhancer sequences that regulate gene activity during various biological stages. Our findings position the Generator as a vital resource for genomic research and biotechnological advancement. By enhancing our ability to interpret and predict genomic sequences, the Generator paves the way for profound improvements in our understanding of complex biological systems and the development of precise genomic interventions. For more technical details, please refer to our paper "GENERator: A Long-Context Generative Genomic Foundation Model".
Data Preparation
For training the Generator (GENERator-eukaryote-1.2b-base), we explored two data processing strategies:
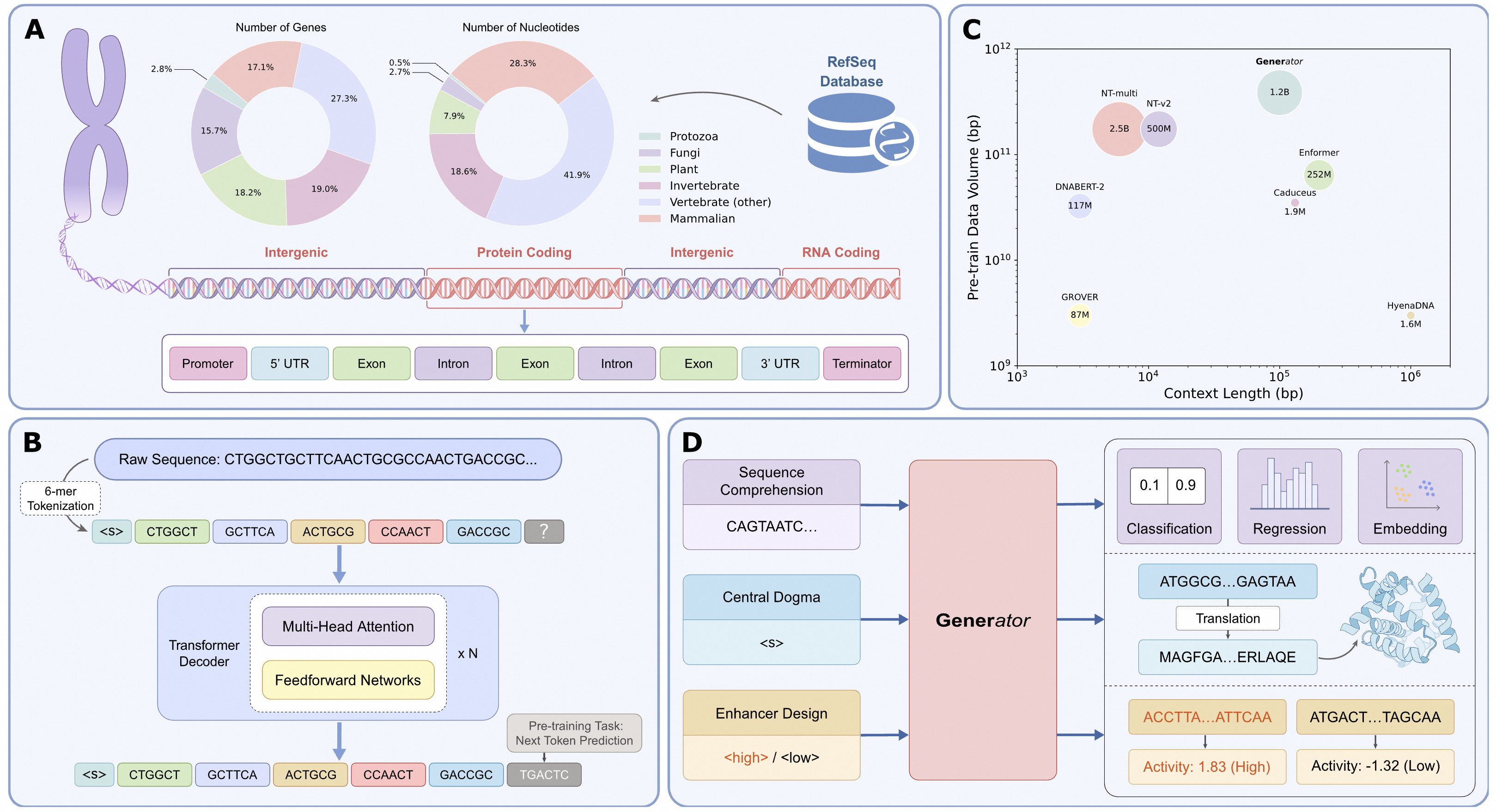
Gene Sequence Training: Utilizing the rich annotation data available in RefSeq, we isolated gene regions from genomic sequences. These regions encompass a wide array of functionalities, including transcription into various RNA molecules, translation into complex proteins, and regulatory functions such as promoters and enhancers that control gene expression. Defined broadly as gene regions, these biologically functional DNA segments formed our training samples, totaling 386B nucleotides.
Whole Sequence Training: In this approach, we fed a mixture of gene and non-gene DNA sequences from all eukaryotic organisms in RefSeq directly into the language model for training. This dataset includes approximately 2T nucleotides. This strategy aligns with the conventional pre-training paradigm for general LLMs and forms the foundational training approach seen in existing DNA language models.
Generally, while Scheme 2 displays a lower pre-training loss, Scheme 1 consistently outperforms Scheme 2 across a range of downstream tasks—even for tasks where non-gene context dominates. One possible explanation for this counterintuitive result is the inherent difference between DNA and human language. DNA is not a concise language but is filled with randomness and redundancy. Over billions of years of evolution, random processes have given rise to a limited number of biologically functional sequences, which represent the "semantics" of DNA. In contrast, the majority of non-gene regions largely consist of segments that have not yet acquired function (or semantic meaning). These regions often include highly repetitive and simple segments, such as long stretches of "AAAAAA" or "GCGCGC", contributing to a lower pre-training loss.
Tokenization
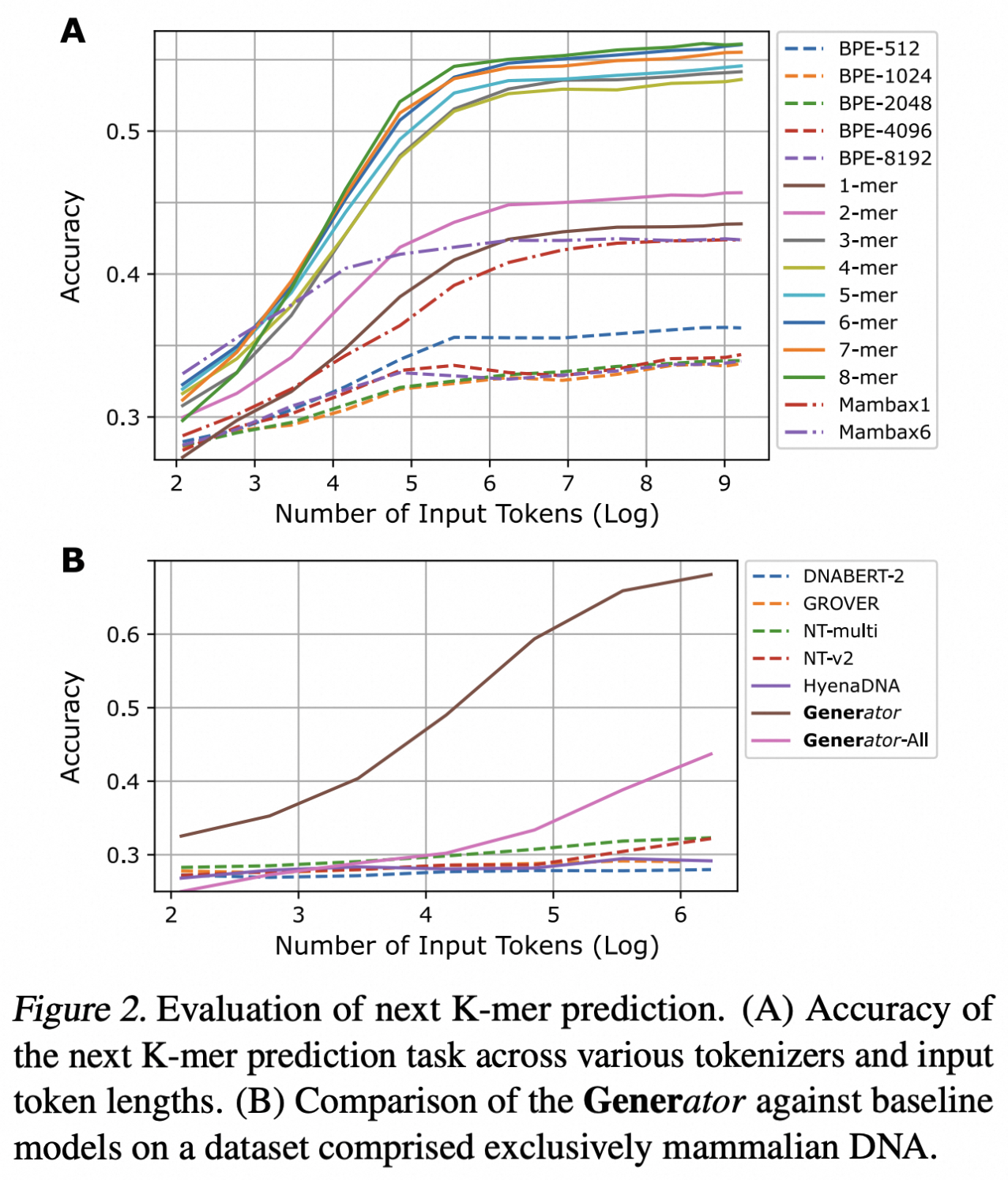
The selection of an appropriate tokenizer involves a trade-off between sequence resolution and contextual coverage. The single nucleotide tokenizer provides the highest resolution, capturing details at the finest level, but for a fixed number of tokens, this increased granularity results in a reduced context window compared to K-mer or BPE tokenizers. Additionally, the computational cost associated with the attention mechanism increases quadratically with sequence length.
Earlier studies such as DNABERT-2 and GROVER demonstrated that the BPE tokenizer is optimal for masked language models. However, our experiments indicate that in the causal language model context, the K-mer tokenizer (6-mer) significantly outperforms other approaches in next token prediction (NTP) pre-training. This observation further underscores the intrinsic differences between DNA sequences and human language, which lack clearly defined lexical boundaries.
Experiments
Benchmark Evaluations
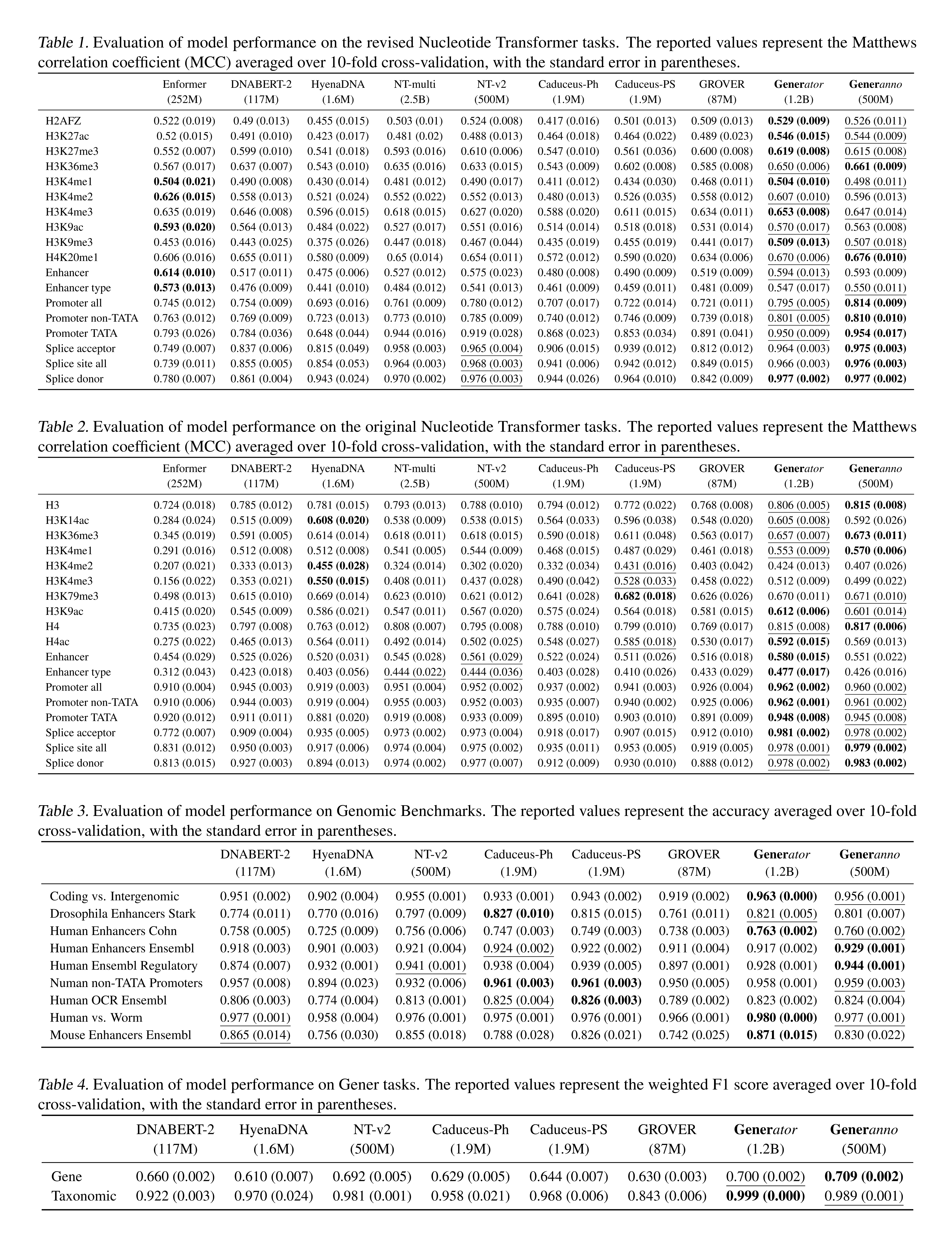
Both Gener models, Generator and Generanno, significantly outperform baseline models across a wide range of benchmarks, establishing them as the top genomic foundation models in the field (2025-02).
Central Dogma
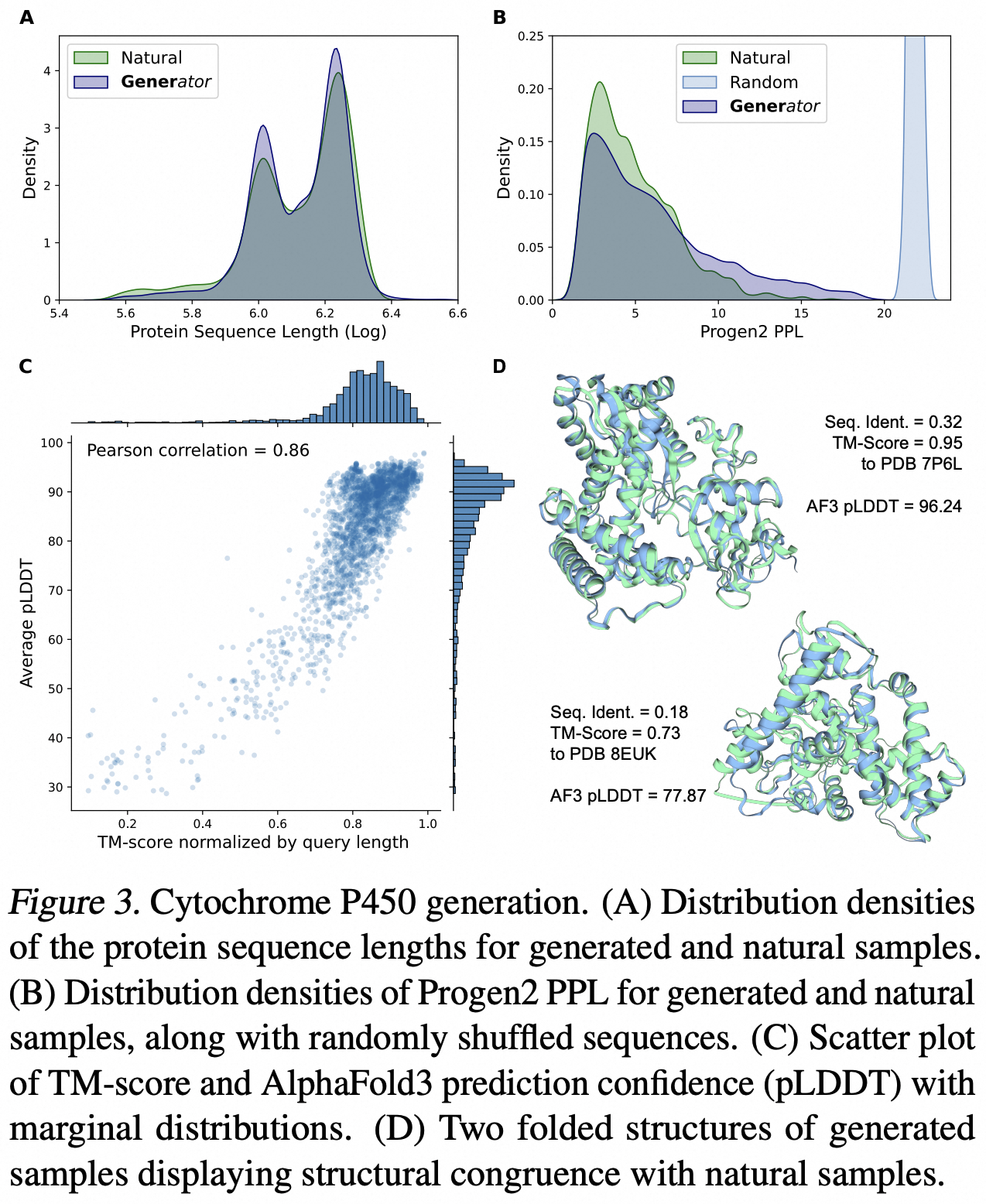
We fine-tuned the Generator model to generate protein-coding DNA sequences. The distribution of the generated sequences closely resembles that of the natural family (Cytochrome P450). We further assessed whether protein language models "recognize" these generated protein sequences by calculating their perplexity (PPL) using ProGen2. The results show that the PPL distribution of generated sequences closely matches that of natural families and significantly differs from that of shuffled sequences. Furthermore, we used AlphaFold3 to predict the folded structures of the generated protein sequences and employed Foldseek to find analogous proteins in the Protein Data Bank (PDB). Remarkably, we identified numerous instances where the conformations of the generated sequences exhibited high similarity to established structures.
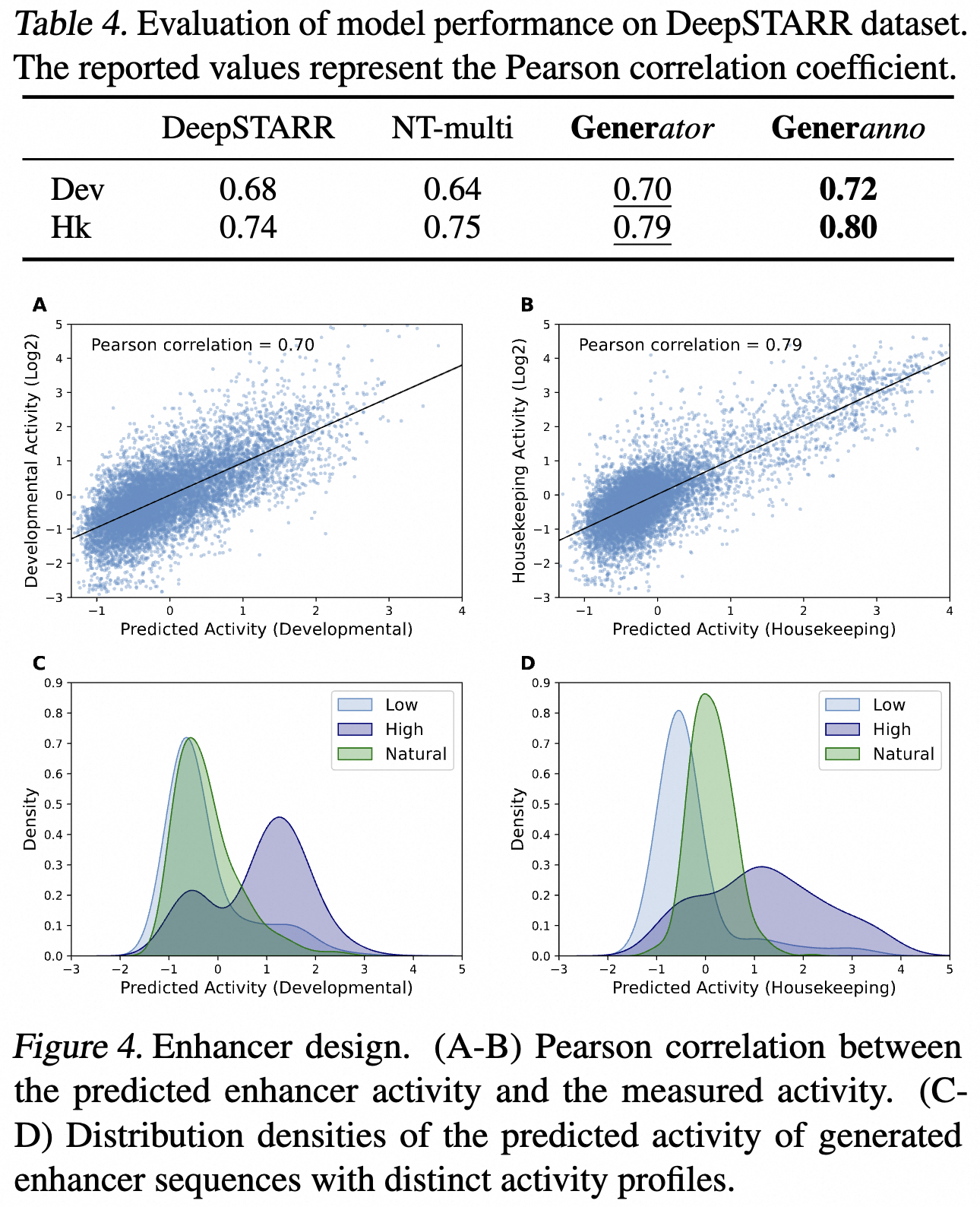
Enhancer Design
We developed an enhancer activity predictor by fine-tuning the Generator, employing enhancer activity data from DeepSTARR. This predictor surpasses the accuracy of DeepSTARR and NT-multi, establishing itself as the current state-of-the-art predictor. By selecting enhancer sequences from the top and bottom quartiles of activity values and labeling them with the prompts <high> and <low>, our refined fine-tuning approach enables the design of enhancer sequences with desired activity profiles. The predicted activities of these sequences exhibit significant differentiation compared to natural samples.